
The Clinical Data Interchange Standards Consortium (CDISC) creates standards that is now mandatory for the regulatory submission to the FDA and PMDA. Study Data Tabulation Model (SDTM) is one of the standards which provides a standard for streamlined data in collection, management, analysis and reporting. If your data is not in SDTM standards then a pharmaceutical company or Clinical Research Organizations (CRO) will regard SDTM mapping to the latest version of SDTM standards for regulatory submission.
SDTM can support data aggregation and warehousing as well as data mining and reuse which are all important to clinical programming for the analysis of clinical trial data when attempting to prove the efficacy and safety of any Investigational New Drug Application (INDA). SDTM is also used in non-clinical data (SEND), medical devices and pharmacogenomics/genetics studies.
This document covers below topics:
- Background on CDISC SDTM Standards
- Advantages of SDTM
- Latest CDISC SDTM Standards and Implementation Guides
- Demonstrate SDTM Mapping
- Medical Devices Implementation Guide
- SDTM Compliance
Background
CDISC has always recognized the need for the development of data standards to improve the process of electronic submissions and exchange of clinical trials information for the benefit of the pharmaceutical industry. SDTM, initially known as Submission Data Model (SDM), was developed by the CDISC Submission Data Standards (SDS) Team in 2004. The SDS Team is comprised of members from different sections of the pharmaceutical industry such as pharmaceutical companies and CROs. The SDS Team meets regularly to review SDTM standards, to continuously improve standards to meet industry requirements. These meetings are also attended by key members from the FDA. CDISC SDTM standards are rapidly evolving with the release of various Therapeutic Area (TA) specific standards as well as regulatory input and requirements.
SDTM Advantages
One interesting question that has come up in the past was, "If it is not yet compulsory to submit data in CDISC SDTM format then why should we change our standards to do this?". Although for FDA and PMDA submissions this is now a requirement, it is clear from listening to other people’s experiences why should we follow SDTM standards. Let’s go through some advantages of SDTM to better understand this.
The main advantage of SDTM is to enable the industry to maintain and follow the same consistent standards for tabulation datasets across all studies. SDTM provides a uniform standard from study to study to ease data exchange internally and across vendors. The CDISC SDTM model is reviewed by many experts across the industry on an on-going basis to strive towards the best standard and to continuously improve the model. Having standardized data means that pharmaceutical companies should be able to collaborate on joint efforts more easily, and in the case of acquisitions, the clinical trials data from the company being acquired should be easy to integrate. The standards are also moving beyond the traditional drug reviews, also covering medical devices. Standardization between studies within a company will also provide more efficiency for the individual company, in terms of standardized code to produce domains and for reporting and analyses. This is an area that Quanticate is focussing on and it can save a lot of time in creation and validation of domains. Having a standardized structure will also make it easier to check the integrity of data from a data management perspective. Standardized checks could be written and performed while a study is still active. There are many such tools that have leveraged this over the years to perform validation, which we will cover later.
Another advantage of SDTM is in the actual review of the data. SDTM datasets follow standard dataset structures and variable attributes which makes it easier for regulatory bodies such as the FDA and PMDA to review data. Regulatory bodies have developed standardized tools for performing checks on submission data which significantly reduce review time and ensuring a basic level of quality before further review is undertaken. Reviewers can be trained on the data standards and standard software tools, so they are able to work more effectively with less preparation time. In fact, the FDA has their own reviewer tools that are run to obtain the information they require to review a submission. It is not necessary to provide subject listings with the submission which would result in fewer questions and faster drug approval time. Similar tools could be used internally by pharmaceutical companies and CROs to help with the review of safety and efficacy of study drugs. For example, standard tools can be developed which can create patient profiles. Hence, one could run a simple report to present the data for a subject (e.g. adverse events, concomitant medications or abnormal clinical laboratory results, electrocardiogram [ECG] or vital sign parameters) in such a way that is consistent, easy to interpret and could show whether the subject was receiving treatment when a particular adverse event or abnormal laboratory result occurred.

SDTM Implementation Guide: Human Clinical Trials
SDTM Implementation guide provides guidance on implementation of core SDTM standards and provides structure of various domains. If collected data is having relevant domain provided in the implementation guide, then this data should be mapped to the domain provided in the implementation guide. If collected data does not have relevant domain in the implementation guide then this data should be mapped to custom domains created based on classes defined in the core SDTM model.
SDTM Domain Classes
SDTM domains are classified into the classes as shown in the image below.
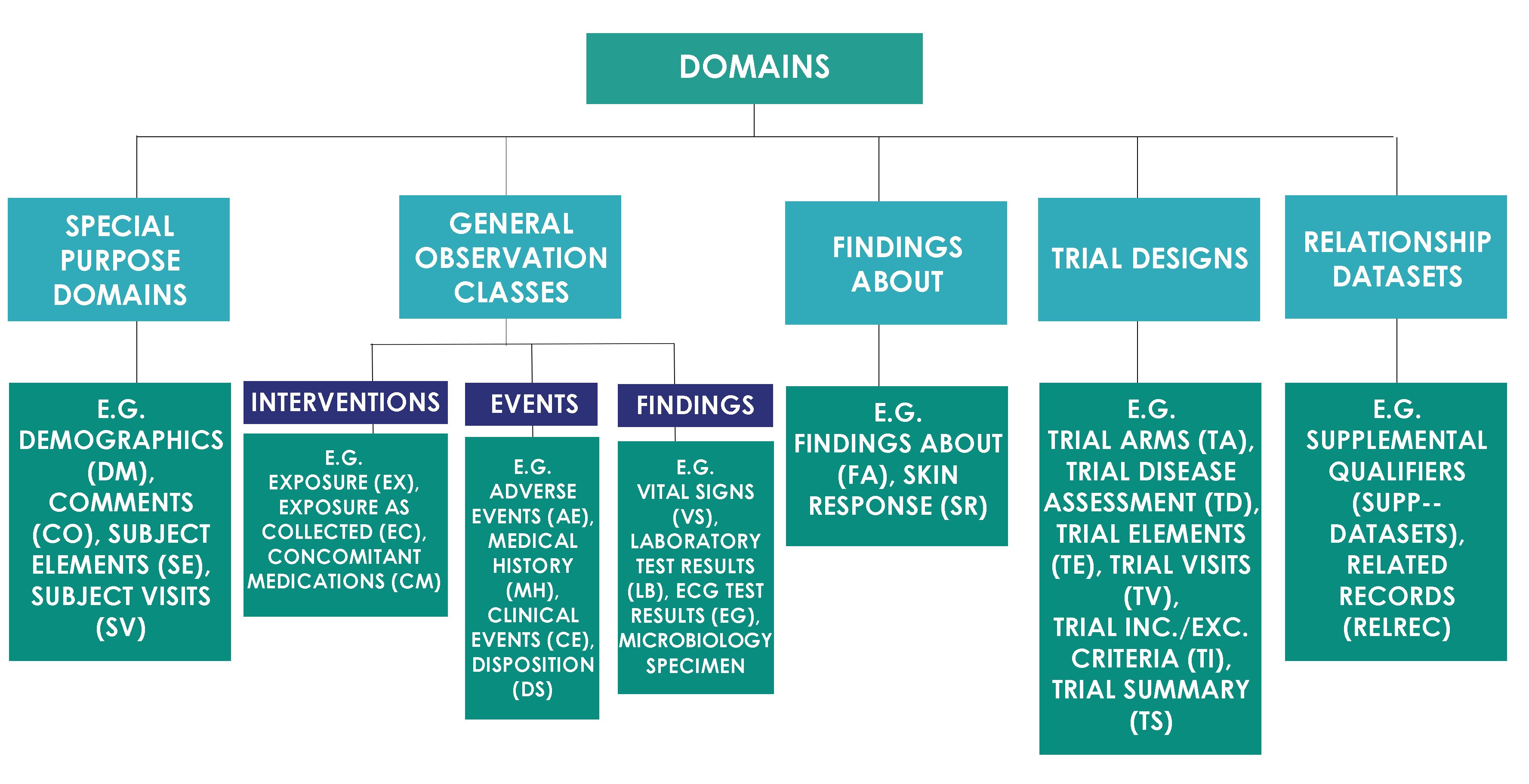
Variable Role
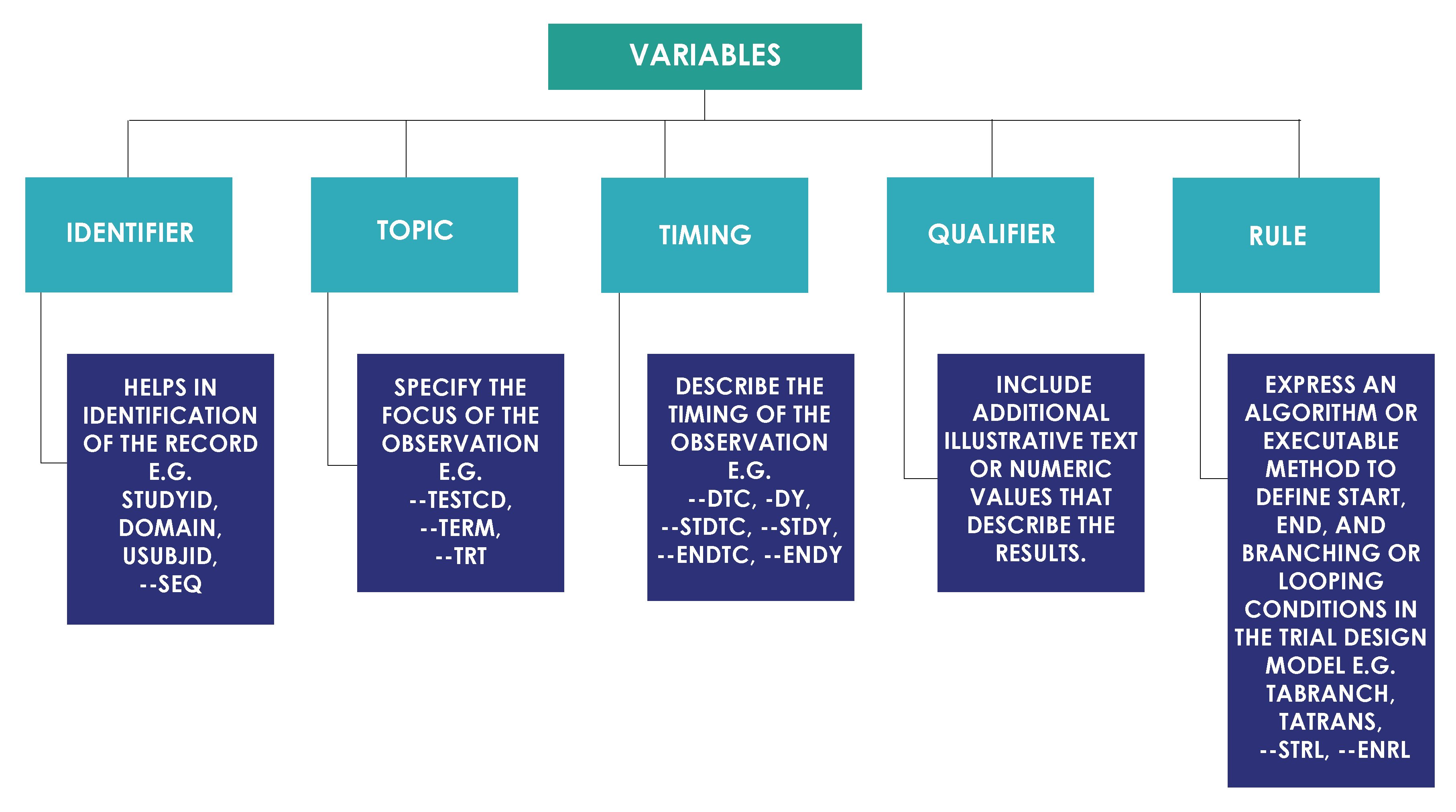
Each domain contains subject's data values collected during clinical trial that are organized as a table of observations (rows) and variables (columns). Each domain can have a list of potential variables. All variables based on their roles are classified into categories as shown in the image below.
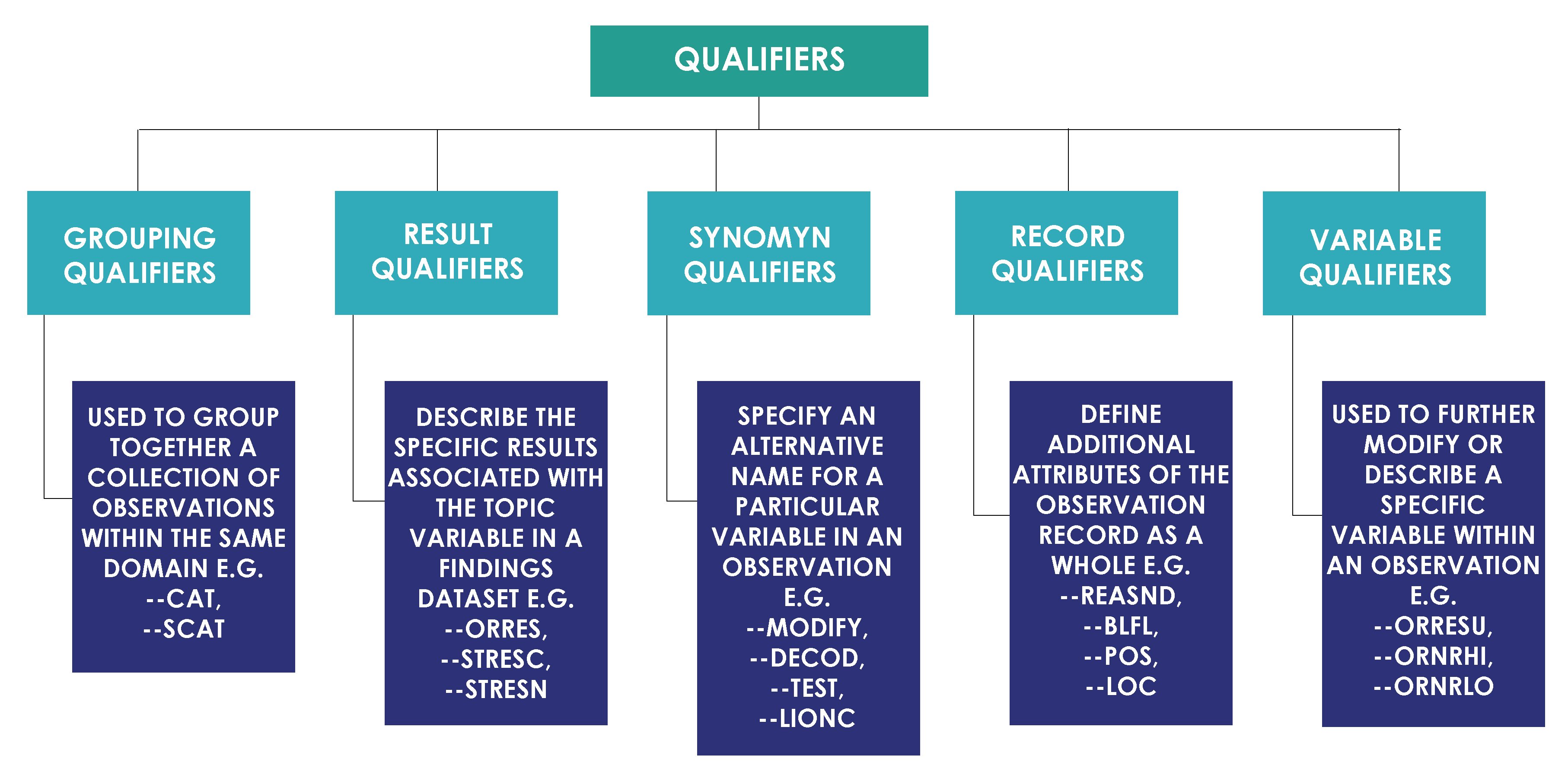
Qualifier variables based on their usage are further classified as shown in the image below.
CDISC Core Variables
There is a concept of core variables in the CDISC in order to control variables with missing values in the submissions. CDISC SDS Team categorizes SDTM variables as below.
- Required Variables: Required variables must always be included in the dataset and cannot be null for any record.
- Expected Variables: Expected variables may contain some null values, but in most cases will not contain null values for every record. When no data has been collected for an expected variable, however, a null column must still be included in the dataset, and a comment must be included in the define.xml to state that data was not collected.
- Permissible Variables: Permissible variable should be used in a domain as appropriate when collected or derived. The Sponsor can decide whether a Permissible variable should be included as a column when all values for that variable are null. The sponsor does not have the discretion to exclude permissible variables when they contain data.
SDTM Mapping
SDTM mapping is a process of converting raw dataset to SDTM domains. Mapping generally follows process as described below.
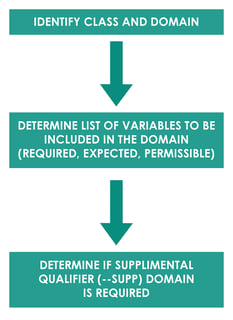
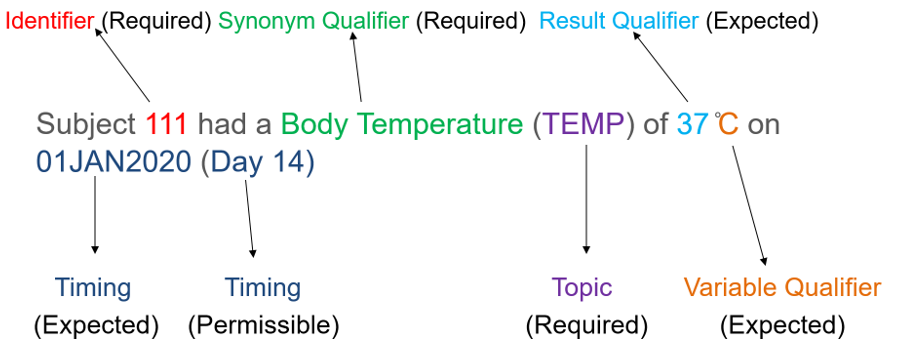
For example, Subject 111 had a Body Temperature (TEMP) of 37 ̊C on 01JAN2020 Day 14. Domain for this observation would be vital signs “VS”. The another Identifier variable is the subject identifier “111”. Topic variable value is the vital sign test code for body temperature “TEMP” and synonym qualifier value is “TEMPERATURE”. Result qualifier value is “37” and variable qualifier value for the result is “̊C”. Timing variable is the study date and day of assessment i.e. 01JAN2020 and Day 14. In this example, all the collected data fit into the standard SDTM variables so –SUPP domain is not required.
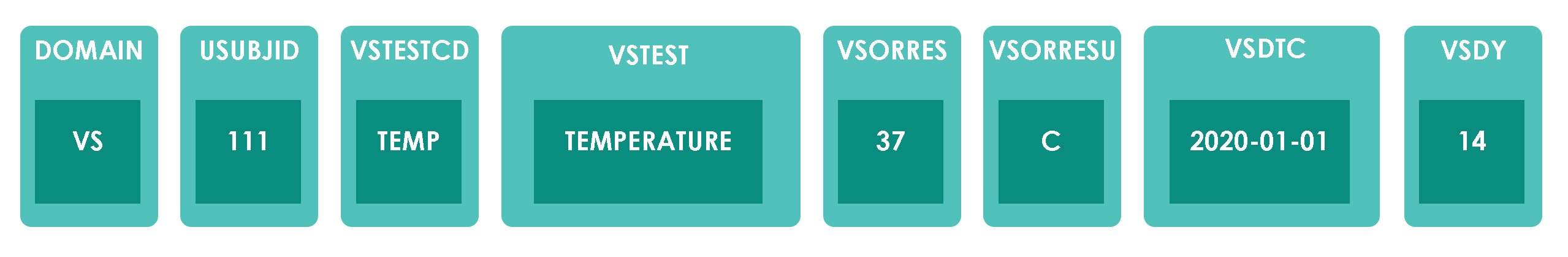
Based on splitting observation into variables, dataset structure would look like below. Data structure shown here is for example purpose only and does not contain all required and expected variables for VS domain.
SDTM Implementation Guide for Medical Devices
Medical devices are any instruments, apparatus or materials to be used in human for diagnosis, prevention and treatment of the disease. Devices are an important and growing part of the medical world, both on their own and in combination with drugs or biologic agents. Medical devices vary both in their intended use and indications for use e.g. low risk devices like tongue depressors, medical thermometers, bedpans, etc. and high-risk devices like implants, prostheses, etc. Medical Device Implementation Guide to the Study Data Tabulation Model defines recommended standards for the submission of data from clinical trials in which medical devices were used. This implementation guide is based on the original SDTM Implementation Guide (SDTMIG) developed for human clinical trials.
Domains per SDTMIG-MD are classified as shown in the image below.
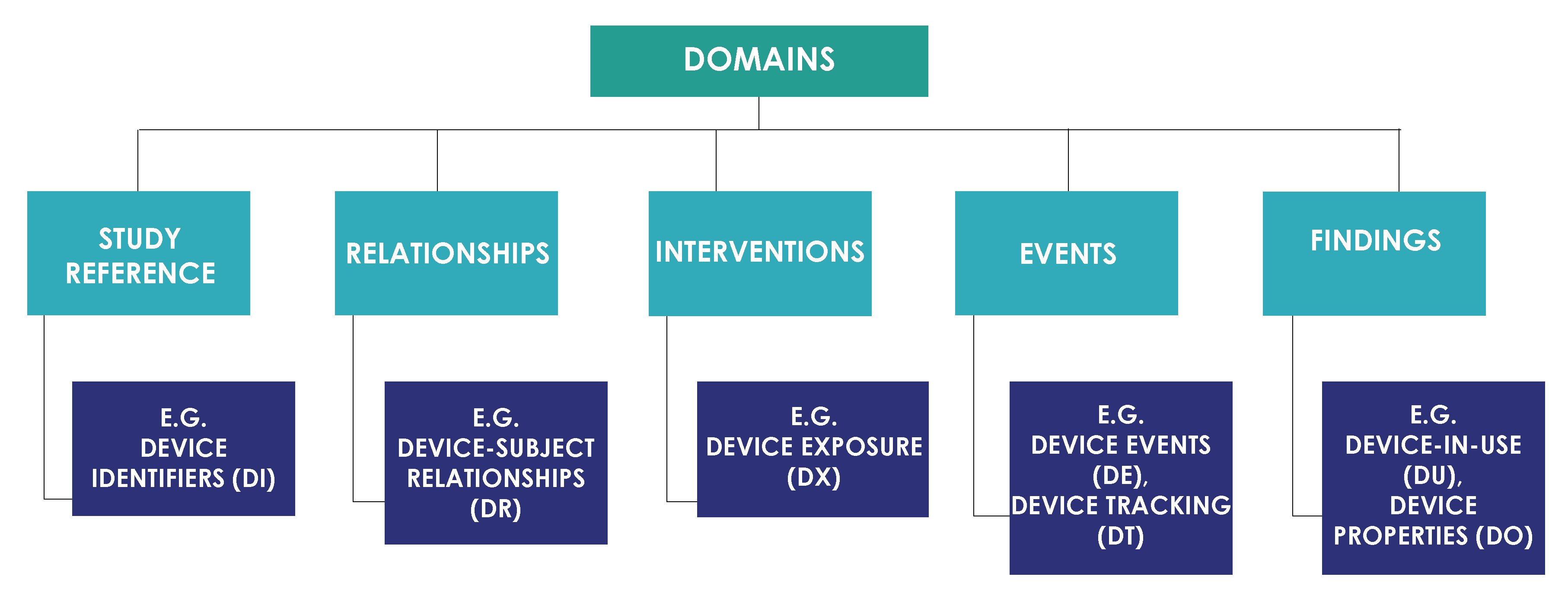
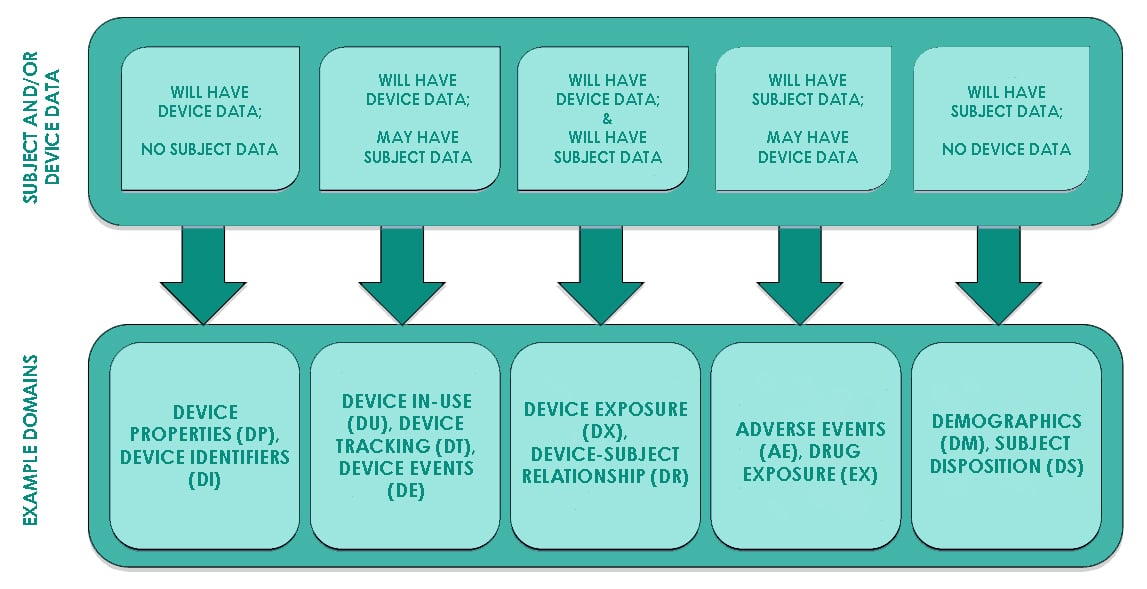
Below image from the SDTMIG-MD explains the relationship of medical device domains with existing SDTMIG domains.
Device Identifiers (DI)
Device Identifiers (DI) is a Study Reference domain that provides a mechanism for using multiple identifiers to create a single identifier for each device. This domain provides primary device identifier variable (SPDEVID) for linking data across device domains. This is separated from the Device Properties (DO) domain because DI contains the total set of characteristics necessary for device identification, whereas DO contains information important for submission but that are not part of the device identifier.
| STUDYID | DOMAIN | SPDEVID | DIPARMCD | DIPARM | DIVAL |
| MD | DI | PC-001 | MANUF | Manufacturer | Ops Limited |
| MD | DI | PC-001 | MODEL | Model | 2000 |
| MD | DI | PC-001 | SERIAL | Serial Number | 001 |
| MD | DI | PC-001 | DEVTYPE | Type of Device | Pacemaker |
Device Properties (DO)
Device Properties (DO) is used to report characteristics of the device that are important to include in the submission and do not vary over the course of the study but are not used to identify the device e.g. shelf life, physical properties, etc.
| STUDYID | DOMAIN | USUBJID | SPDEVID | DOTESTCD | DOTEST | DOORRES | DORRESU |
| MD | DO | MD-999-001 | PC-001 | COMPOS | Composition | Titanium | |
| MD | DO | MD-999-001 | PC-001 | SHELLIFE | Shelf Life | 6 | Years |
| MD | DO | MD-999-001 | PC-001 | PWEIGHT | Pacemaker Weight | 50 | gms |
Device Exposure (DX)
The Device Exposure (DX) domain records the details of a subject’s direct interaction or contact with a medical device or the output from a medical device, usually but not always the device under study.
| STUDYID | USUBJID | SPDEVID | DXTRT | DXLOC | DXLAT | DXSTDTC | DXENDTC |
| MD | MD-999-001 | PC-001 | Pacemaker | ATRIUM | RIGHT | 2018-02-05 |
Device-In-Use (DU)
Device-In-Use (DU) contains the values of measurements and settings that are intentionally set on a device when it is used, and may vary from subject to subject or other target. These are characteristics that exist for the device, and have a specific setting for a use instance. DU is distinct from Device Properties (DO), which describes static characteristics of the device.
Device Tracking and Disposition (DT)
Device Tracking and Disposition (DT) represents a record of tracking events for a given device (e.g., initial shipment, deployment, return, destruction). Different events would be relevant to different types of devices. The last record represents the device disposition at the time of submission.
| STUDYID | DOMAIN | SPDEVID | DTTERM | DTPARTY | DTPRTYID | DTSTDTC |
| MD | DT | PC-001 | SHIPPED | SITE | 999 | 2018-02-02 |
| MD | DT | PC-001 | IMPLANTED | SUBJECT | 001 | 2018-02-05 |
Device Events (DE)
Device Events (DE) contains information about various kinds of device-related events, such as device malfunctions. A device event may or may not be associated with a subject or a visit. If a device event, such as a malfunction, results in an adverse event, then this information should be recorded in the Adverse Events (AE) domain. The relationship between the AE and a device malfunction in DE can be recorded using RELREC domain.
Device-Subject Relationships (DR)
Device-Subject Relationships (DR) links each subject to devices to which they have been exposed. This domain provides a single, consistent location to find the relationship between a subject and a device, regardless of the device or the domain in which subject-related data may have been collected or submitted.
| STUDYID | USUBJID | SPDEVID |
| MD | MD-999-001 | PC-001 |
SDTM Compliance
Compliance checks of CDISC SDTM data are also an important step in validating the conversion of raw datasets to SDTM datasets. Compliance checks of SDTM datasets can be done using open source validation tools like PROC CDISC in SAS, Pinnacle 21 Validator (previously known as OpenCDISC Validator) or WebSDM to name a few, but there are new tools being developed all of the time and thorough validation is seen as a clear objective to increase quality while reducing time for development and review cycles.
Another question that often appears is, "Is it possible to submit data as CDISC SDTM compliant if those data fail a particular requirement?" The data should follow CDISC SDTM standards unless there is a very good reason to deviate from the standard guidelines. However, it may not always be possible to follow standard guidelines exactly e.g. CDISC controlled terminology for adverse event severity expects AESEV variable to have values like MILD, MODERATE or SEVERE but if adverse event severity is collected on CRF as "Yes" or "No" and CRF updates are not possible then this does not conform to CDISC controlled terminology. Please note seriousness of adverse event is mapped to AESER and this example discusses about adverse event severity which is mapped to AESEV. When this SDTM is run thorough validation checks for CDISC compliance through some of the tools mentioned previously, this would result in an error. In such cases, it is acceptable to submit the data because it may just mean that a particular report for AESEV may not be able to run. However, detailed explanations of errors along with reasons for not being able to fix the errors should be added in the Study Data Reviewer's Guide (SDRG). However, as more companies and CRF designers become familiar with CDISC standards, this is now less of an issue than when CDISC was new to the industry. However, the fact remains that it is possible to submit data that fails specific checks, but this should be a last resort and a valid reason should be given in the SDRG with the submission.
Conclusion
While the detailed CDISC standards provide additional details, the fact remains that data standardization allows for interoperability of data between analytical tools and organizations. CDISC standards are developed to enable the pharmaceutical industry to standardize the clinical trial data analysis process and benefit the mankind with the new drug discoveries with reduced review time.

Related Blog Posts:
- Why do a 3rd of Regulatory Submissions fail the Technical Rejection Criteria?
- Using CDISC SDTM to Improve Cost and Quality in Integrated Summaries
Subscribe to the Blog
FOLLOW QUANTICATE ON:


© 2024 Quanticate